Spondylometaphyseal dysplasia, Schmidt type
spon-dy-lo-me-ta-fy-see-al dis-PLAY-zha, shmidt type
Also known as: SMD Schmidt type, Schmidt type SMD
At a Glance
What is Spondylometaphyseal dysplasia, Schmidt type?
Spondylometaphyseal dysplasia, Schmidt type, is a rare genetic disorder that affects bone development. It primarily impacts the spine and the metaphyses of long bones. This condition is caused by mutations in specific genes that are crucial for bone growth and development. Over time, individuals with this disorder may experience progressive skeletal deformities. Early symptoms often include short stature and abnormal curvature of the spine, while later symptoms can involve joint pain and limited mobility. Early diagnosis is critical to manage symptoms and improve quality of life. The condition can place a significant emotional and financial burden on families due to the need for ongoing medical care. Prognosis varies, but many individuals lead active lives with appropriate management. Daily life may include regular physical therapy and orthopedic interventions. Affected individuals often require adaptive devices to assist with mobility. Social and educational support is essential to help them achieve their full potential. Genetic counseling is recommended for families to understand the inheritance pattern and recurrence risks.
Medical Definition
Spondylometaphyseal dysplasia, Schmidt type, is a genetic skeletal disorder characterized by abnormalities in the spine and metaphyseal regions of long bones. Pathologically, it involves defective endochondral ossification due to mutations affecting collagen or other bone matrix proteins. Histological findings typically reveal disorganized growth plate cartilage and abnormal bone remodeling. It is classified under the group of spondylometaphyseal dysplasias, which are further subdivided based on genetic and clinical features. Epidemiologically, it is an extremely rare condition with an estimated prevalence of 1 in 1,000,000. The disease course involves progressive skeletal deformities that can lead to functional limitations if not managed appropriately.
Spondylometaphyseal dysplasia, Schmidt type
Spondylometaphyseal dysplasia, Schmidt type Symptoms
Symptoms vary in severity between individuals. Early diagnosis and management can significantly improve outcomes.
Very Common
Short stature manifests as a significantly reduced height compared to peers of the same age and sex. This is caused by abnormalities in bone growth and development due to genetic mutations affecting collagen production. Over time, the growth rate remains below average, leading to a noticeable height difference by adolescence. Daily life can be affected by challenges in physical activities and social interactions, but growth hormone therapy and supportive care can help manage these issues.
Skeletal abnormalities present as irregular bone shapes, particularly in the spine and long bones. These are due to disruptions in the normal ossification process, often linked to collagen defects. As the child grows, these abnormalities can become more pronounced, potentially leading to joint pain and mobility issues. Physical therapy and orthopedic interventions can help improve function and quality of life.
Joint pain is experienced as discomfort or aching in the joints, often exacerbated by physical activity. It is caused by abnormal joint structures and cartilage wear due to improper bone alignment. The pain may increase with age and can limit participation in sports and other activities. Pain management strategies, including medication and physical therapy, can help alleviate symptoms.
Common
Spinal deformities such as scoliosis or kyphosis appear as abnormal curvatures of the spine. These occur due to uneven growth and development of vertebral bones. As the child grows, these deformities can worsen, potentially affecting posture and respiratory function. Regular monitoring and, in some cases, surgical intervention may be necessary to manage these issues.
Delayed motor development is observed as a lag in reaching movement milestones like walking or running. This delay is attributed to musculoskeletal abnormalities and reduced muscle strength. Over time, children may catch up to some extent, but they might continue to experience coordination challenges. Early intervention with physical therapy can support motor skill development.
Vision problems can include nearsightedness or other refractive errors, presenting as difficulty seeing clearly. These issues arise from structural abnormalities in the eye, often linked to collagen defects. Vision may deteriorate over time, affecting academic performance and daily activities. Regular eye examinations and corrective lenses can help manage these symptoms.
Less Common
Hearing loss may manifest as difficulty hearing sounds or understanding speech. It is caused by structural anomalies in the ear or auditory pathways, potentially linked to genetic factors. The degree of hearing loss can vary and may progress with age. Hearing aids and other assistive devices can improve hearing and communication.
Dental abnormalities, such as malocclusion or delayed tooth eruption, are noticeable in the alignment and timing of teeth development. These occur due to irregularities in jaw and tooth structure. As the child grows, dental issues can lead to difficulties in chewing and speech. Orthodontic treatment and regular dental care are essential for managing these conditions.
What Causes Spondylometaphyseal dysplasia, Schmidt type?
Spondylometaphyseal dysplasia, Schmidt type, is primarily caused by mutations in the COL2A1 gene located on chromosome 12q13.11. The COL2A1 gene encodes the alpha-1 chain of type II collagen, a crucial protein for the normal development of cartilage and vitreous humor in the eye. Mutations in COL2A1 can lead to the production of abnormal type II collagen, which disrupts the protein's triple-helix structure and stability. This structural disruption impairs the assembly and secretion of collagen fibrils, leading to defective cartilage matrix formation. The defective cartilage matrix results in abnormal bone growth and skeletal dysplasia, affecting the spine and metaphyses of long bones. The cellular stress from misfolded proteins can trigger an unfolded protein response, leading to apoptosis of chondrocytes. This cellular apoptosis contributes to the degeneration of cartilage and bone tissue, causing skeletal abnormalities. In some cases, neuroinflammation may occur due to the release of inflammatory cytokines from stressed cells, potentially affecting neural tissues. The degeneration of cartilage and bone structures can lead to joint pain and limited mobility, characteristic symptoms of the condition. The pattern of symptoms, such as short stature and spinal deformities, arises from the specific roles of type II collagen in skeletal development. Variability in disease severity among patients may be due to the location and type of mutation within the COL2A1 gene, as well as potential modifier genes or environmental factors. Additionally, the presence of other genetic variations can influence the phenotype, contributing to the heterogeneity of clinical presentations. The immune response may exacerbate tissue damage, further influencing the progression and severity of symptoms. Understanding the molecular mechanisms underlying these mutations provides insights into potential therapeutic targets for managing the condition.
How is Spondylometaphyseal dysplasia, Schmidt type Diagnosed?
Typical age of diagnosis: Spondylometaphyseal dysplasia, Schmidt type, is typically diagnosed in early childhood when growth abnormalities become apparent. Parents often notice delayed milestones and disproportionate short stature, prompting medical evaluation. Diagnosis is usually confirmed by a combination of clinical, radiological, and genetic assessments. Early diagnosis is crucial for managing symptoms and planning appropriate interventions.
The clinician looks for disproportionate short stature, limb deformities, and spinal abnormalities. A detailed family history is taken to identify any hereditary patterns. Physical examination may reveal joint laxity, scoliosis, and metaphyseal irregularities. This step helps to narrow down the differential diagnosis and guides further testing.
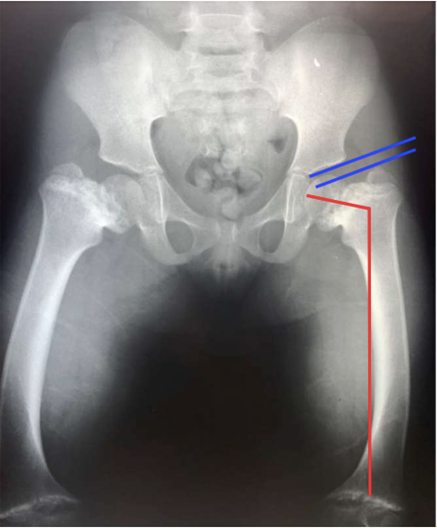
X-rays are the primary imaging modality used to assess bone structure. Specific abnormalities such as metaphyseal irregularities and vertebral changes are visible. These findings help confirm the diagnosis of spondylometaphyseal dysplasia. Imaging also helps exclude other skeletal dysplasias with similar presentations.
Routine laboratory tests may include serum calcium, phosphate, and alkaline phosphatase levels. These tests help rule out metabolic bone diseases. Abnormal results may show altered bone turnover markers. The results guide the clinician in deciding whether genetic testing is warranted.
Genetic testing involves sequencing the COL2A1 gene. Mutations such as missense or splice site mutations are commonly found. These results confirm the diagnosis of spondylometaphyseal dysplasia, Schmidt type. Genetic results also provide information for family counseling and future family planning.
Spondylometaphyseal dysplasia, Schmidt type Treatment Options
Bisphosphonates are used to manage bone pain and prevent fractures. They work by inhibiting osteoclast-mediated bone resorption. Specific drugs like alendronate and pamidronate are commonly used. Clinical evidence shows improved bone density and reduced fracture rates. Side effects may include gastrointestinal discomfort and rare cases of osteonecrosis of the jaw.
Techniques include stretching, strengthening exercises, and gait training. The goal is to improve mobility, strength, and functional independence. Sessions are typically conducted 2-3 times a week for several months. Measurable outcomes include improved range of motion and reduced pain. Long-term benefits include enhanced quality of life and reduced disability.
Surgery is indicated for severe limb deformities affecting function. The procedure involves cutting and realigning bones to correct deformities. Expected benefits include improved limb alignment and function. Surgical risks include infection, nerve damage, and non-union. Post-operative care requires physical therapy and regular follow-up.
The care team includes orthopedic specialists, physical therapists, and genetic counselors. Interventions focus on managing symptoms and improving quality of life. Psychosocial support strategies involve counseling and support groups. Family education is essential for understanding the condition and managing expectations. Long-term monitoring includes regular assessments and adjustments to the care plan.
When to See a Doctor for Spondylometaphyseal dysplasia, Schmidt type
- Severe difficulty breathing — this could indicate a serious respiratory complication requiring immediate medical attention.
- Sudden loss of mobility — may suggest a spinal cord issue or severe joint problem needing urgent evaluation.
- Acute chest pain — could be a sign of cardiac involvement or other serious conditions that need emergency care.
- Persistent joint pain — may indicate worsening of skeletal issues; consult a doctor for assessment and management.
- Noticeable spinal curvature — could suggest progression of skeletal deformities; seek orthopedic evaluation.
- Delayed growth in children — may reflect underlying metabolic or genetic issues; a pediatrician should be consulted.
- Mild back discomfort — monitor for changes in intensity or frequency and manage with home remedies.
- Occasional joint stiffness — keep track of any patterns and try gentle exercises to maintain mobility.
Spondylometaphyseal dysplasia, Schmidt type — Frequently Asked Questions
Is this condition hereditary?
Spondylometaphyseal dysplasia, Schmidt type, is typically inherited in an autosomal dominant pattern. This means there is a 50% chance of passing it to offspring if one parent is affected. De novo mutations can occur, meaning the condition can appear in individuals with no family history. Carrier status is not applicable as it is not a recessive condition. Genetic counseling is recommended for affected families to understand inheritance patterns and reproductive options.
What is the life expectancy for someone with this condition?
Life expectancy can vary depending on the severity and age of onset of symptoms. Early intervention and management of complications can improve outcomes. Mortality is often related to respiratory issues or severe skeletal complications. Treatment can enhance quality of life but may not significantly extend lifespan. Realistic expectations should focus on managing symptoms and maintaining function.
How is this condition diagnosed and how long does diagnosis take?
Diagnosis involves clinical evaluation, genetic testing, and imaging studies such as X-rays. The process from first symptoms to diagnosis can take several months due to the rarity of the condition. Specialists such as geneticists, orthopedists, and radiologists are often consulted. Delays in diagnosis are common due to symptom overlap with other skeletal dysplasias. Confirmation is typically achieved through genetic testing identifying specific mutations.
Are there any new treatments or clinical trials available?
Current research is exploring gene therapy and novel pharmacological approaches. ClinicalTrials.gov is a resource for finding ongoing trials related to this condition. Patients should discuss potential trial participation with their healthcare provider. The timeline for new treatments becoming widely available can be several years. Staying informed about research developments is crucial for accessing emerging therapies.
How does this condition affect daily life and activities?
Mobility may be significantly impacted, requiring assistive devices for walking. Educational accommodations may be necessary due to physical limitations. Social and emotional challenges include coping with chronic pain and potential isolation. Family burden can be high due to caregiving demands and medical appointments. Supportive therapies and adaptive equipment can greatly enhance daily functioning and quality of life.
Learn More
Support & Resources
References
Content generated with support from peer-reviewed literature via PubMed.
- 1.COL2A1 Mutation in Spondylometaphyseal Dysplasia Algerian Type.
Matsubayashi S, Ikema M, Ninomiya Y et al. · Mol Syndromol · 2013 · PMID: 23653587
- 2.Development of therapies for rare genetic disorders of GPX4: roadmap and opportunities.
Cheff DM, Muotri AR, Stockwell BR et al. · Orphanet J Rare Dis · 2021 · PMID: 34688299
- 3.Spondylometaphyseal dysplasia, east-African type: a new form of early, severe SMD with rounded vertebrae.
Verloes A, Lepage P, Baumann C et al. · Am J Med Genet · 2002 · PMID: 12457408
This content is for educational purposes only and is not a substitute for professional medical advice, diagnosis, or treatment.Last reviewed: 2026-05-05